введение
Синдром Окамо́то-Клайн (АКС), также называемый синдромом Окамо́то, это синдром множественных врожденных пороков развития, связанный с интеллектуальной недостаточностью. Он в основном вызывается вариантами потери функции в гене HNRNPK, который кодирует гетерогенный ядерный рибонуклеопротеин K (hnRNP K). Это можно классифицировать как очень редкое аутосомно-доминантное генетическое состояние.
Синдром Окамо́то впервые был описан в 1997 году Нобухико Окамо́то и др. в Японии после наблюдения схожих симптомов и физических черт у двух неродственных японских младенцев.
Синдром Окамо́то-Клайн впервые был описан в 2015 году Пин-И Билли Ау, Антони Д. Клайн и др. после обнаружения мутаций в HNRNPK у двух пациентов с аналогичными симптомами в практиках в Калгари, Альберта, Канада, и Балтиморе, Мэриленд, США. Обе стороны подали ген как кандидата на онлайн-сервис GeneMatcher, который связал их вместе и позволил подтвердить синдром.
Окамо́то в 2019 году предложил, что синдром Окамо́то-Клайн и синдром Окамо́то являются синонимами из-за мутации в гене HNRNPK, так как симптомы практически идентичны[1][2].
клинически значимая анатомия
Существует белок под названием гетерогенный ядерный рибонуклеопротеин K (hnRNP K), который является частью гена HNRNPK и связывается с ДНК или ее РНК и другими белками. Собирая определённые белки вместе с ДНК или РНК, этот белок помогает контролировать активность генов и производство белков. Регулируя определённые гены и белки, hnRNP K играет роль в нормальном развитии и функционировании нескольких систем организма, включая мозг[2].
этиология
АКС вызывается мутациями в гене HNRNPK, расположенном на хромосоме 9 в позиции q21.32, что приводит к производству мало или вовсе не производящему hnRNP K белку. Эта измененная генетическая активность и производство белков нарушают нормальное развитие или функционирование нескольких систем организма. Это также может влиять на развитие мозга, приводя к интеллектуальной недостаточности, задержкам развития и другим неврологическим проблемам у людей с этим состоянием.
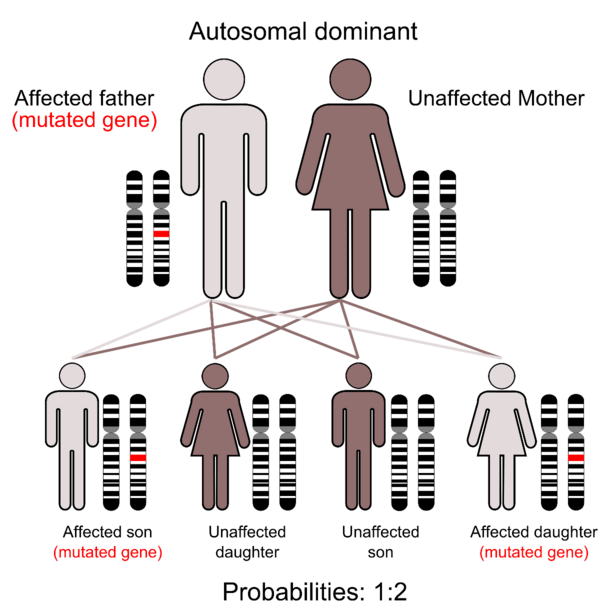
Это генетически наследуется по аутосомно-доминантному образцу. Большинство случаев возникает из-за новых (de novo) мутаций в гене, которые происходят во время формирования репродуктивных клеток (яйцеклеток или сперматозоидов) у родителя пострадавшего индивида или в раннем эмбриональном развитии. Эти случаи встречаются у людей без семейного анамнеза этого расстройства.
клиническая картина
Это состояние характеризуется:
- Врожденный гидронефроз,
- Низкий мышечный тонус и сниженные рефлексы,
- Пороки сердца: такие как стеноз аортального клапана, предсердный или желудочковый септальные дефекты, двустворчатый аортальный клапан или открытый артериальный проток.
- Интеллектуальная недостаточность,
- Характерные черты лица: заметные, опущенные уши, открытый, опущенный рот и опущенные веки (птоз)
- Неврологические и скелетные аномалии,
- Инфекции мочевыводящих путей,
- Речевые и двигательные навыки,
- Снижение роста: малый вес и размер.[3]
диагностика
Состояние может быть диагностировано по физическим симптомам и подтверждено генетическим тестированием. Генетические тесты могут быть проведены:
- секвенированием всего экзома;
- сравнительной геномной гибридизацией (для микроделеции).
Секвенирование Сэнгера может подтвердить характер мутации [4].
управление/вмешательства
Управление зависит от симптомов. Физиотерапевтическое вмешательство может быть предложено для низкого тонуса и функционального роста. Реабилитационная команда должна включать команду кардиологов, неврологов, физиотерапевтов, эрготерапевтов, специальных педагогов, логопедов и других специалистов, связанных с симптомами.
Генетическое консультирование является важным аспектом управления этим состоянием. Генетические тесты до рождения возможны для беременностей с повышенным риском, если известен патогенный вариант HNRNPK в семье.
Прогноз состояния еще неясен из-за небольшого количества диагностированных случаев[4].
рекомендации
- ↑ Au PYB, You J, Caluseriu O, Schwartzentruber J, Majewski J, Bernier FP, и др. GeneMatcher помогает идентифицировать новый синдром пороков развития с интеллектуальной недостаточностью, уникальными чертами лица и аномалиями скелета и соединительной ткани, вызванными новыми вариантами в HNRNPK. Hum Mutat. 2015;36:1009–14. doi: 10.1002/humu.22837.
- ↑ 2.0 2.1 Okamoto, N. "Синдром Окамо́то имеет общие черты с синдромом Окамо́то-Клайн и вызван мутацией HNRNPK". American Journal of Medical Genetics. Part A. Май 2019, 179 (5): 822–826. doi:10.1002/ajmg.a.61079. ISSN 1552-4833
- ↑ Au, P; Innes, M; Kline,A.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Au-Kline Syndrome", GeneReviews®, University of Washington, Seattle, 2019, PMID 30998304
- ↑ 4.0 4.1 Au, P. Y., Goedhart, C; Ferguson, M; Breckpot, J; Devriendt, K; Wierenga, K; и др. "Фенотипический спектр синдрома Окамо́то-Клайн: шесть новых случаев и обзор литературы". European Journal of Human Genetics. Сент. 2018. 26 (9): 1272–1281. doi:10.1038/s41431-018-0187-2. ISSN 1476-5438. PMC 6117294. PMID 29904177