Описание
Мышечная дистрофия Эмери-Дрейфусс (Muscular Dystrophy) (EDMD) — это редкое генетическое дегенеративное заболевание, поражающее скелетные мышцы и сердце. EDMD является одним из девяти типов мышечной дистрофии.[1]
EDMD можно подразделить на 3 категории:
• X-сцепленный EDMD
• Аутосомно-доминантный EDMD
• Аутосомно-рецессивный EDMD
Распространенность
В Соединенных Штатах около 250 000 человек страдают каким-либо типом мышечной дистрофии.[2]
Предполагается, что X-сцепленная мышечная дистрофия Эмери-Дрейфусс поражает 1 из 100 000 человек в общей популяции[3][4], и считается третьим по распространенности типом мышечной дистрофии.[2] X-сцепленный EDMD полностью выражен только у мужчин; однако у 10-20% носителей X-сцепленного EDMD среди женщин развиваются дефекты сердечной проводимости и/или мышечная слабость.[2]
Частота аутосомно-доминантного EDMD неизвестна. Аутосомно-рецессивный тип расстройства является очень редким, с только несколькими зарегистрированными случаями по всему миру.[4] Аутосомные типы EDMD поражают мужчин и женщин в равной степени.[2]
Этиология/Причины
Гены:
X-сцепленный EDMD:
Этот тип EDMD вызывается мутацией в гене EMD, который участвует в создании белка эмирина. Эмирин необходим для нормального функционирования скелетных и сердечных мышц; хотя не ясно, как недостаток эмирина вызывает признаки и симптомы, характерные для этого заболевания.[5][6]
Аутосомно-доминантный и рецессивный EDMD:
Эти EDMD вызваны мутацией в гене LMNA, который помогает в создании белков ламина A и ламина C. Мутация создает измененную версию этих белков.[5][6]
Существует теория, что мутация в этих генах вызывает дестабилизацию ядерной мембраны, что может привести к разрушению мышц. До сих пор не понятно, почему поражаются только скелетные и сердечные мышцы, несмотря на присутствие белков по всему телу.[1]
Наследование EDMD:
X-сцепленный EDMD: мутация гена EMD передается по X-хромосоме. Мужчины более склонны к этому расстройству и наследуют мутировавший ген от матери-носителя. Женщина должна иметь две мутировавшие X-хромосомы для развития этого расстройства. Женщина с только одной мутировавшей X-хромосомой считается носителем расстройства. Носительница обычно бессимптомна, но все еще подвержена риску сердечных проблем.[6]
Аутосомно-доминантный EDMD: это означает, что одна копия измененного гена LMNA достаточна, чтобы вызвать расстройство. Не обязательно наличие семейной истории EDMD, чтобы эта мутация гена возникла.[6]
Аутосомно-рецессивный EDMD: возникает, когда у человека есть две копии измененного гена LMNA, по одной от каждого родителя. Человек с только одной мутацией гена считается носителем.[6]
Характеристики/Клиническая картина
EDMD характеризуется тремя признаками:
• Ранние контрактуры (ахиллово сухожилие, локти и задние шейные мышцы)
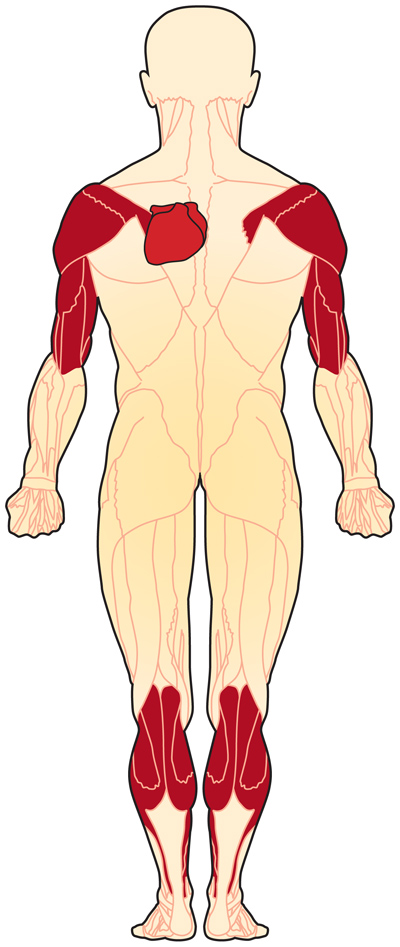
• Прогрессирующая атрофия по плечо-малоберцовому распределению (проксимальные мышцы верхней конечности и дистальные мышцы нижней конечности). Позднее в ходе заболевания слабеют и проксимальные поясные мышцы конечностей.
• Дефекты сердечной проводимости[7]
Признаки и симптомы:
• Симметричная слабость бицепсов и трицепсов при сохранении дельтовидных мышц
• Слабость лица, бедра и рук встречается редко и проявляется позднее в ходе заболевания
• Ходьба на носочках
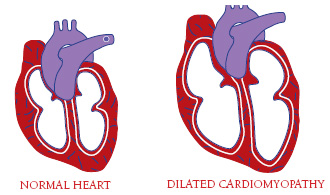
• Кардиомиопатия (может привести к сердечной недостаточности)
• Аtriовентрикулярный блок
• Паралич предсердий
• Внезапная смерть[5]
• Обмороки вследствие воздействия на сердце[1]
Симптомы часто проявляются к 10 годам
Сердечные аномалии обычно обнаруживаются к 20 годам
Женщины, являющиеся носителями X-сцепленного EDMD, не имеют вовлеченности скелетных мышц, но могут иметь сердечные аномалии.[1]
Связанные сопутствующие заболевания
Лица с EDMD имеют широкий круг сердечных аномалий, включая остановку предсердий, фибрилляцию предсердий, застойную сердечную недостаточность и кардиоэмболический инсульт. Популяция подвержена высокому риску развития тяжелой брадикардии, что несет риск внезапной смерти, и суправентрикулярных тахиаритмий, которые ассоциированы с высоким риском тромбоэмболического инсульта. Важно отметить, что сердечное и мышечное поражение не были показаны как тесно связанные, и случаются случаи тяжелой кардиомиопатии у пациентов с только легкими мышечными симптомами. Возможен такой вариант, когда инсульт выступает первым клиническим проявлением EDMD.[8]
Системное вовлечение
Мышечная дистрофия Эмери-Дрейфусс поражает произвольные мышцы, а также сердце. Ранние симптомы включают слабость и атрофию по плечо-малоберцовому распределению, которое со временем затрагивает мышцы плечевого и тазового пояса.[9] Контрактуры возникают и могут усложнить движения рук, шеи, лодыжек и позвоночника, приводя к "ходьбе на носочках" и затруднению сгибания локтей.[1]
Дефициты сердечной проводимости и аритмии могут возникнуть, приводя к дильатированной кардиомиопатии, плохой переносимости нагрузок, брадикардии, обморокам, застойной сердечной недостаточности и повышенному риску инсульта и внезапной смерти.[4][3]
Женщины, являющиеся генетическими носителями X-сцепленного EDMD, также могут быть подвержены риску сердечных проблем, причем риск увеличивается с возрастом. Однако носители обычно не проявляют мышечной слабости или контрактур.[1]
Диагностические тесты/Лабораторные тесты/Лабораторные значения
Клинический диагноз[9]
Клинический диагноз EDMD ставится на основе наличия триады, указанной в разделе Клиническая картина.
Другие неспецифические клинические находки[9]
- Электромиограмма (ЭМГ) обычно показывает миопатические черты с нормальными исследованиями проводимости нервов. Однако, обнаружены нейропатические паттерны при сцепленном с Х-хромосомой EDMD и аутосомно-доминантном EDMD.
- КТ показывает диффузный паттерн вовлечения мышц, включая бицепс, камбаловидную мышцу, малоберцовую, внешние части четырехглавой мышцы бедра, ягодичные и паравертебральные мышцы. Находки в области икр и задней части бедра были отмечены у пациентов с аутосомно-доминантным EDMD.
Неспецифические лабораторные находки[9]
- Концентрация CK в сыворотке может быть в норме или умеренно повышена в 2-20 раз выше верхней границы нормы. Повышения чаще наблюдаются в начале заболевания, чем на поздних стадиях.
- Гистопатология мышц показывает неспецифические миопатические или дистрофические изменения: изменения в размере волокон, увеличение внутренних ядер, увеличение эндомизиальной соединительной ткани, некротические волокна. Электронная микроскопия может показать специфические изменения в ядерной архитектуре. Биопсия мышц редко проводится в диагностических целях из-за недостатка специфичности.
Иммунодетекция эмерина (обнаруживается с помощью иммунофлуоресценции и/или вестерн-блота): отсутствует у 95% людей со сцепленным с Х-хромосомой EDMD. Обычно выражена у людей с аутосомно-доминантным EDMD. Иммунодетекция FHL1 (обнаруживается с помощью иммунофлуоресценции и/или вестерн-блота): отсутствует или значительно снижена у людей с X-связным EDMD, ассоциированным с FHL1.
Генетическое тестирование
Генетическое тестирование может определить наличие определённых дефектов, вызывающих EDMD, и помочь предсказать течение болезни, а также оценить риск передачи заболевания следующему поколению.[1]
Дифференциальный диагноз
Мышечная дистрофия Эмери-Дрейфуса должна быть дифференцирована от других диагнозов, в первую очередь от других типов мышечной дистрофии.
- Мышечная дистрофия Дюшенна - впервые распознаётся в возрасте от 3 до 6 лет. Характеризуется мышечной слабостью и атрофией, начинающейся в области таза, прогрессирующей к плечам и в конечном счёте поражающей большинство основных мышц тела.[2]
- Мышечная дистрофия Беккера - начинается во второй или третьей декаде жизни. Практически исключительно поражает мужчин. Ослабление мышц бедра и плеч, развивается ненормальная походка и возможно лёгкое умственное отставание.[2]
- Фациоскапулогумеральная мышечная дистрофия (мышечная дистрофия Ландузи-Дежерина) - начало обычно в подростковом возрасте или раннем взрослом возрасте. Характеризуется слабостью мышц лица, плеч и верхней части руки. Возможны нарушения в закрытии глаз, движении губ и поднятии рук над головой. В конце концов, слабость и атрофия могут поразить и нижние конечности.[2]
- Поясная мышечная дистрофия - группа редких прогрессирующих генетических расстройств, характеризующихся атрофией и слабостью поясов бедра и плеч.[2]
- Синдром ригидного позвоночника - редкое нервно-мышечное расстройство, характеризующееся гипотонией, слабостью, контрактурами и атрофией мышц. Часто возникает сколиоз. Может наблюдаться в сочетании с EDMD.[2]
- Миопатии - к рассмотрению могут быть отнесены болезнь Помпе, дерматомиозит, полимиозит и немалиновая миопатия.[2]
- Миастения - аутоиммунное расстройство периферических нервов, характеризующееся слабостью при повторяющемся использовании мышцы, за которым следует восстановление в период отдыха. Наиболее часто поражает бульбарные мышцы с колеблющейся общей слабостью.[10]
- Спинальная мышечная атрофия - проявляется слабостью и атрофией конечностей, дыхательных и бульбарных мышц.[10]
Для дифференциации EDMD от неврологической проблемы может быть проведён неврологический скрининг.
Клинические признаки верхнего мотонейрона[11]:
- Слабость или паралич
- Спастичность
- Повышенные сухожильные рефлексы
- Знак Бабинского
- Потеря поверхностных брюшных рефлексов
- Незначительная или отсутствующая атрофия мышц
Клинические признаки нижнего мотонейрона[11]:
- Слабость или паралич
- Истощение и фасцикуляции вовлечённых мышц
- Гипотония или вялость
- Потеря сухожильных рефлексов
- Нормальные брюшные рефлексы и отрицательный знак Бабинского
Неврологические проблемы также могут проявляться сенсорными дефицитами.
Медицинское управление
Специфического лечения для ЭДМД не существует. Лечение определяется на основе индивидуальных симптомов.
Генетическое консультирование может понадобиться для пациента и его/ее семьи.[2]
Лекарства
Антиаритмические препараты для нарушения АВ
Антитромбоэмболические препараты для предотвращения церебральной тромбоэмболии кардиогенного происхождения
Препараты, которых следует избегать:
• Деполяризующие релаксанты мышц (например, сукцинилхолин)
• Летучие анестетики (например, галотан, изофлуран)[2]
Лечение:
• Хирургическое вмешательство:
Устранение контрактур и лечение сколиоза [9]
(Тем не менее, контрактуры, вероятно, рецидивируют)[1]
Пересадка сердца может понадобиться при терминальной сердечной недостаточности[9]
• Физиотерапия[1]
• Сердечный кардиостимулятор или имплантируемый кардиовертер-дефибриллятор для
блокады АВ и нарушения проводимости[9]
• Фармакологическая и нефаракологическая терапия сердечной недостаточности[9]
Мониторинг:
• Ежегодный контроль сердца (включая ЭКГ и эхокардиографию)[9]
• Респираторная функция[9]
Будущие исследования:
В настоящее время не существует эффективного лечения мышечной дистрофии. Однако проводится исследование генетически ориентированной терапии для лечения мышечной дистрофии Дюшенна. Это, возможно, в дальнейшем приведет к варианту лечения ЭДМД.[12]
Управление физиотерапией
Физиотерапия для пациентов с мышечной дистрофией Эмери-Дрейфусса сосредоточена на лечении нарушений, вызванных заболеванием. Пациенты должны обращаться к физиотерапевтам вскоре после постановки диагноза. Из-за более позднего возраста постановки диагноза по сравнению с другими мышечными дистрофиями, этой группе, скорее всего, не нужно оценивать их двигательную функцию. Тем не менее, физиотерапевтические вмешательства будут иметь большое значение для качества жизни пациентов[13].
- Пассивное и активное растяжение для увеличения гибкости и предотвращения контрактур
- Укрепляющие упражнения для поддержания и улучшения мышечной силы
- Респираторные техники, такие как ассистированное кашляние, тренировка дыхательных мышц[14]
- Активизация: Технические средства такие как трости, бандажи и инвалидные коляски могут потребоваться для поддержания мобильности
Отчеты о случаях / Исследования случаев
Мышечная дистрофия Эмери-Дрейфусса: отчет о случае [15]
Трансплантация сердца у пациентов с мышечной дистрофией Эмери-Дрейфусса: отчеты о случаях[16]
Необычный вариант редкого типа мышечной дистрофии [17]
Ресурсы
Ресурсы для пациентов и семей, страдающих мышечной дистрофией Эмери-Дрейфусса:
Справочник Национальной библиотеки медицины по генетике
Кампания против мышечной дистрофии
НИЗ/Национальный институт неврологических расстройств и инсульта
Информационный центр по генетическим и редким заболеваниям (GARD)
Лечение CMD (врожденной мышечной дистрофии)
Ссылки
- ↑ 1.0 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 Hauptmann-Thanheuser. Эмери-Дрейфус мышечная дистрофия: Диагностика. http://mda.org/disease/emery-dreifuss-muscular-dystrophy/diagnosis (дата обращения 3 марта 2014)
- ↑ 2.00 2.01 2.02 2.03 2.04 2.05 2.06 2.07 2.08 2.09 2.10 2.11 2.12 Lammerding, J. Эмери-Дрейфус мышечная дистрофия. Доступно по адресу: http://www.rarediseases.org/rare-disease-information/rare-diseases/byID/590/viewFullReport. Дата доступа 3 марта 2014. (дата обращения 3 марта 2014).
- ↑ 3.0 3.1 Helbling-Leclerc A, Bonne G, Schwartz K. Эмери-Дрейфус мышечная дистрофия. http://www.nature.com/ejhg/journal/v10/n3/full/5200744a.html (дата обращения 3 марта 2014).
- ↑ 4.0 4.1 4.2Мышечная дистрофия Эмери-Дрейфусса. http://ghr.nlm.nih.gov/condition/emery-dreifuss-muscular-dystrophy (дата обращения: 3 марта 2014 года).
- ↑ 5.0 5.1 5.2 Rocha CT, Hoffman EP. Поясная и врожденная мышечная дистрофии: современные диагностика, управление и передовые технологии. Текущие отчеты по неврологии и нейронаукам. 2010 июль;10(4):267-76. PubMed PMID: 20467841.http://www.ncbi.nlm.nih.gov/books/NBK1436/
- ↑ 6.0 6.1 6.2 6.3 6.4 Мышечная дистрофия Эмери-Дрейфусса. http://ghr.nlm.nih.gov/condition/emery-dreifuss-muscular-dystrophy (дата обращения: 16 марта 2014 года).
- ↑ Emery AE. Мышечная дистрофия Эмери-Дрейфусса — ретроспектива 40 лет. Невромускульные нарушения. 2000;10(4):228-32.
- ↑ Boriani G, Gallina M, Merlini L, и др. Клиническая значимость фибрилляции/трепетания предсердий, инсульта, имплантации кардиостимулятора и сердечной недостаточности при мышечной дистрофии Эмери-Дрейфусса: долгосрочное лонгитудинальное исследование. Инсульт. 2003;34 (4):901–908. Доступно по адресу: http://stroke.ahajournals.org/content/34/4/901.abstract.
- ↑ 9.0 9.1 9.2 9.3 9.4 9.5 9.6 9.7 9.8 9.9 Bonne G, Leturcq F, Yaou RB. Мышечная дистрофия Эмери-Дрейфусса. http://www.ncbi.nlm.nih.gov/books/NBK1436/ (дата обращения: 3 марта 2014 года)
- ↑ 10.0 10.1 Lopate, G. Дифференциальная диагностика мышечной дистрофии Эмери-Дрейфусса. http://emedicine.medscape.com/article/1178994-differential (дата обращения: 6 марта 20)
- ↑ 11.0 11.1 Greenberg DA, Aminoff MJ, Simon RP. Клиническая неврология, 8-е издание. Соединенные Штаты Америки: The McGraw-Hill Companies, Inc; 2012.
- ↑ Goyenvalle A, Seto JT, Davies KE, Chamberlain J. Терапевтические подходы к мышечной дистрофии. Генетика человека и молекулярная биология. 2011;20(R1):R69-R78.
- ↑ 13.0 13.1 Руководство физиотерапевта по мышечным дистрофиям у детей. http://www.moveforwardpt.com/symptomsconditionsdetail.aspx?cid=11b647e2-58e0-4b84-9d27-61d24ff3af10#top (дата обращения: 7 апреля 2014 года).
- ↑ Bonne G, Leturcq F, Ben Yaou R. Мышечная дистрофия Эмери-Дрейфусса. GeneReviews® [Интернет]. 2004.
- ↑ JSaraiva F, Rodrigues D, Andrade H, Negrão L, Gonçalves L, Marinho A, Providência LA. Мышечная дистрофия Эмери-Дрейфусса: клинический случай. Revista Portuguesa de Cardiologia (Английское издание). 2012 мар 1;31(3):241-5.
- ↑ Dell’Amore A, Botta L, Suarez SM, Forte AL, Mikus E, Camurri N, Ortelli L, Arpesella G. Пересадка сердца у пациентов с мышечной дистрофией Эмери-Дрейфусса. В Proceeding трансплантации 2007 дек 1 (Том 39, № 10, рр. 3538-3540). Elsevier.
- ↑ Jayashankar CA, Somasekar DS, Gurucharan A, Perugu PK, Ashwath SV, Santosh KV. Необычный вариант редкого типа мышечной дистрофии. Международный журнал исследований в области медицинских наук. 2013 июл;1(3):313.