Введение
Мышечная дистрофия Беккера (МДБ)—одна из группы X-сцепленных мышечных дистрофий, проявляет такую же схему вовлечения мышц, как и мышечная дистрофия Дюшенна (МДД), но с более медленным прогрессом клинического течения[1], и потеря способности к ходьбе отмечается в большинстве случаев МДБ примерно в возрасте 37 лет[2].
Клинически релевантная анатомия
Дистрофин отвечает за связь цитоскелета каждой мышечной фибры с подлежащей базальной мембраной. Отсутствие дистрофина препятствует проникновению кальция в клеточную мембрану, что влияет на сигналы клетки: вода проникает в митохондрии, вызывая разрыв клетки. В результате сложного каскадного процесса, включающего несколько путей, увеличенный окислительный стресс внутри клетки повреждает сарколемму, что приводит к гибели клетки, а мышечные волокна подвергаются некрозу и заменяются соединительной тканью. Тем не менее, переоценка гипотезы о том, что утрата связи базальной мембраны и цитоскелета являлась основным фактором, участвующим в мышечных дистрофиях, была подкреплена открытием синтазы оксида азота (NOS), сигнальных молекул, требующих неповрежденного комплекса дистрофина для связывания с сарколемой, а также тем, что некоторые мышечные дистрофии с вовлечением проксимальных мышц (LGMDs) обусловлены уменьшением саркогликанового комплекса, тогда как комплекс дистрофина на сарколемме не нарушен.[3]
Патологический процесс
МДБ является типом рецессивной, X-сцепленной дистрофинопатии. Клинические вариации у пациентов с МДБ обусловлены различиями в мутациях дистрофина из-за делеций экзонов[4]. Дефект является мутацией в белке, называемом дистрофином, расположенного на хромосоме Xp21.2, и может быть унаследован как X-сцепленный рецессивный признак. Пациенты без явной X-сцепленной схемы наследования могут иметь дефекты в других генах, влияющих на ассоциированные с дистрофином гликопротеины.[5].
Уровень дистрофина при МДБ составляет в среднем 30-80% от нормы, тогда как при МДД он составляет менее 5%[6].
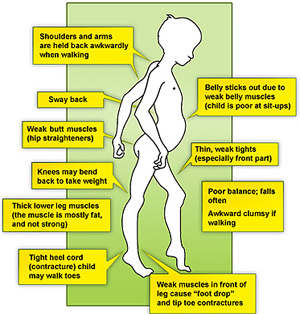
Клиническая картина
- Задержка двигательных вех развития может стать первым наблюдением со стороны родителей.
- Неуклюжесть
- Частые падения
- Трудности при подъеме с пола, может проявляться знак Говерса (неспецифичный)
- Субклинические случаи могут проявляться позже в жизни; дилатационная кардиомиопатия может стать первым признаком МДБ.
- Контрактуры
- Слабость может быть ограничена определенными проксимальными мышцами
- Сохранение силы сгибателей шеи может присутствовать
- «Качелиобразная» походка в тяжелых случаях
Дифференциальная диагностика
- Мышечная дистрофия Дюшенна: Более тяжелая и с ранним началом, чем МДБ. Прогноз неблагоприятный.
- Полимиозит: Отсутствие дистальной псевдогипертрофии помогает отличить его от МДБ.
- Спинальная мышечная атрофия: Отсутствие мутации гена дистрофина при ДНК-пробе провоцирует спинальную мышечную атрофию как альтернативный диагноз.[5]
- Пояснично-конечностная мышечная дистрофия: Это состояние трудно отличить от МДБ; однако псевдогипертрофия икроножной мышцы отсутствует.[5]
- Дилатационная кардиомиопатия: кардиомиопатия является одним из самых серьезных осложнений и основной причиной смертности при дистрофиях. Но дилатационная кардиомиопатия может быть отдельной сущностью с иной генетической этиологией или по другим причинам, не связанных с мышечной дистрофией.[8]
- Мышечная дистрофия Эмери-Дрейфусса: Ранние контрактуры и кардиальные дефекты помогают отличить ее от МДБ. [9]
- Миастения гравис: Флуктуационная слабость скелетных мышц имитирует клиническую картину МДБ, но слабость лицевых мышц, птоз и диплопия часто встречаются.[10]
- Метаболические миопатии: Большинство пациентов жалуются на слабость мышц и боль во время физической активности, а не в покое[11].
Диагностические процедуры
- Уровень креатинкиназы в сыворотке - умеренное или сильное повышение[12]
- Анализ делеций гена дистрофина с помощью нескольких методов, таких как Мультиплексный анализ зондов, зависимый от литирования (MLPA), флуоресцентный анализ in situ (FISH) и полимеразная цепная реакция (PCR). MLPA — наиболее распространенный метод.[13]
- Мышечная биопсия с окрашиванием антителами против дистрофина
- Магнитно-резонансная томография сердца (МРТ)
- Электромиография: показана для дифференциации между первичным заболеванием нервного процесса и миопатией.
- Исследования проводимости нервов - ожидается, что они будут нормальными
- Электрокардиограмма/Экокардиограмма
- Функция легких
- Рентген для выявления каких-либо аномалий костей в результате контрактур и истощения мышц, отслеживания сколиоза
- Тесты функции печени на трансаминазы
Меры исхода
Меры исхода для количественной оценки прогрессирования заболевания, включая:
- 6-минутный тест ходьбы
- Шкала оценки Северная звезда для амбулаторных пациентов,
- Время, необходимое для подъема по четырем ступеням
- Время, необходимое для подъема с пола
- Производительность верхних конечностей.
Одним из ограничений этих мер является тот факт, что они нацелены либо на пациентов, способных ходить, либо на неспособных ходить[14]. Тем не менее, по мере прогрессирования заболевания меры исхода меняются, что затрудняет использование одной меры исхода для анализа пациента. Исследования продолжаются для создания единой меры для мышечных дистрофий.
Медицинское Управление
- Пациентам не предоставляются лекарства для специфического лечения БМД.
- Лекарства назначаются для лечения симптомов, которые обычно сопутствуют БМД (например, сердечные препараты при сердечно-сосудистых заболеваниях).
- Кортикостероидные препараты используются для того, чтобы помочь людям оставаться способными ходить так долго, как это возможно, задерживая воспалительный процесс.[15]
Управление Физиотерапией
- Обучение пациента и его семьи важно в этих случаях.
- Пассивные и активные растяжки для улучшения гибкости суставов (объема движений) и предотвращения или задержки развития контрактур.
- Такие виды деятельности, как езда на велосипеде и плавание, могут использоваться для улучшения сердечно-сосудистой выносливости и силовых тренировок. Однако следует принимать во внимание, чтобы эти действия не были слишком трудоемкими или утомительными, так как они могут нанести больше вреда мышцам.
- Респираторная тренировка - на ранних стадиях состояния физиотерапевт участвует в помощи поддерживать активность ребенка. На более поздних стадиях состояния физиотерапевт также поможет с респираторными проблемами, используя спирометрию, правильное позиционирование, метод хуффинга и эффективного кашля.
- Улучшение двигательных навыков ребенка и помощь ему в достижении вех развития с использованием техник Проприоцептивного Нейромышечного Функционирования|PNF, различных подходов, таких как подход Рудса, Бруннстрем и Бобат
- Прогрессивные упражнения с сопротивлением с минимальными весами без утомления мышц.
- Массаж можно проводить до мышц для уменьшения боли и контрактур.
Трудотерапия
Деятельность повседневной жизни (ADL) может быть модифицирована в зависимости от уровня нарушений. Адаптации с использованием инструментов могут быть выполнены с использованием вспомогательных средств, таких как палочки для одевания, поручни, модифицированные наборы для еды, ручки, приподнятые сиденья для туалета и т. д.[16] Предметы могут быть размещены на более низком уровне для пациентов, прикованных к инвалидному креслу.
Ортезы
Решаются вопросы подвижности, включая необходимость в устройствах для содействия мобильности, таких как скутер или полностью адаптированное инвалидное кресло с индивидуальным сиденьем и спинкой, специальными поддержками и электрическим приводом.
Логопедия
Проблемы дисфагии могут быть оценены логопедом. Клиническая оценка может привести к рекомендации избегать определенных текстур пищи и вязкости жидкостей, а также исключить определенные позы во время кормления.
Рекреационная терапия
Хобби, желания и увлечения могут быть стимулированы для улучшения самочувствия и общего физического состояния человека. Эти действия следует оценивать на основе интересов и способностей ребенка. Различные музыкальные инструменты, танцы, ремесла, искусство, а также йога могут быть изучены или мотивированы в ребенке.
Осложнения
Могут возникнуть осложнения в виде:[17]
- кардиомиопатии, прогрессирующей потери функций легких и печени, потери возможности ходить, когнитивных нарушений и переломов костей.
- высокого риска послеоперационных инфекций грудной клетки.
- Рабдомиолиз, приводящий к миоглобинурии и последующей почечной недостаточности.
- кортикостероиды, вызывающие недостаточность надпочечников и иммуносупрессию.
Ресурсы
Мышечная дистрофия
Дюшенн Мышечная Дистрофия - Исследование Случая
Литература
- ↑ Bushby KM, Thambyayah M, Gardner-Medwin D. Распространенность и частота возникновения мышечной дистрофии Беккера. The Lancet. 1991 Apr 27;337(8748):1022-4.
- ↑ Bushby KM, Gardner-Medwin D. Клинические, генетические и дистрофиновые характеристики мышечной дистрофии Беккера I. Естественная история. Журнал неврологии. 1993; 240:98–104.
- ↑ Duggan DJ, Hoffman EP. Аутосомно-рецессивная мышечная дистрофия и мутации комплекса саркогликана. Невромышечные расстройства. 1996; 6:475–482.
- ↑ Nicolas A, Raguénès-Nicol C, Ben Yaou R, Ameziane-Le Hir S, Chéron A, Vié V, Claustres M, Leturcq F, Delalande O, Hubert JF, Tuffery-Giraud S. Тяжесть мышечной дистрофии Беккера связана с структурой дистрофина. Генетика человека. 2015 Mar 1;24(5):1267-79.
- ↑ 5.0 5.1 5.2 Thada PK, Bhandari J, Umapathi KK. Мышечная дистрофия Беккера. [Обновлено 2021 Aug 1]. В: StatPearls [Интернет]. Трежер-Айленд (FL): StatPearls Publishing; 2021 Jan-. Доступно из: https://www.ncbi.nlm.nih.gov/books/NBK556092/
- ↑ Angelini C, Fanin M, Pegoraro E, и др. Клинически-молекулярная корреляция у 104 пациентов с легкой Х-связанной мышечной дистрофией: характеристика субклинических фенотипов. Невромышечные расстройства. 1994 Jul. 4(4):349-58.
- ↑ Medicosis Perfectionalis. Мышечная дистрофия Беккера. Доступно из: http://www.youtube.com/watch?v=7Ult-apDFB8 [последний доступ 16/4/2021]
- ↑ Dec GW, Fuster V. Идиопатическая дилатационная кардиомиопатия. N Engl J Med. 1994 Dec 08;331(23):1564-75.
- ↑ Puckelwartz M, McNally EM. Мышечная дистрофия Эмери-Дрейфус. Руководство по клинической неврологии. 2011;101:155-66.
- ↑ Vincent A, Palace J, Hilton-Jones D. Миастения. Lancet. 2001 Jun 30;357(9274):2122-8. [PubMed] [Список литературы]
- ↑ Tarnopolsky MA. Метаболические миопатии. Continuum (Minneap Minn). 2016 Dec;22(6, Мышечные и нервно-мышечные соединения):1829-1851
- ↑ Flanigan KM. Мышечные дистрофии Дюшенна и Беккера. Neurol Clin. 2014 Aug;32(3):671-88, viii.
- ↑ Hwa HL, Chang YY, Chen CH, Kao YS, Jong YJ, Chao MC, Ko TM. Мультиплексная амплификация зависимой от связывания проб идентификация делеций и дупликаций гена мышечной дистрофии Дюшенна у тайваньских субъектов. J Formos Med Assoc. 2007 May;106(5):339-46.
- ↑ Меры исходов при мышечной дистрофии Дюшенна: чувствительность к изменениям, клиническая значимость и последствия для клинических испытаний Joana Domingos Francesco Muntoni https://doi.org/10.1111/dmcn.13634
- ↑ Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, Case LE, Clemens PR, Hadjiyannakis S, Pandya S, Street N, Tomezsko J, Wagner KR, Ward LM, Weber DR., Рабочая группа по вопросам ухода при ДМД. Диагностика и лечение мышечной дистрофии Дюшенна, часть 1: диагностика и нейромышечная, реабилитационная, эндокринная и гастроэнтерологическая и нутриционная терапия. Lancet Neurol. 2018 Mar;17(3):251-267.
- ↑ Grootenhuis MA, de Boone J, van der Kooi AJ. Жизнь с мышечной дистрофией: последствия для качества жизни, связанные со здоровьем, для детей и взрослых. Health Qual Life Outcomes. 2007; 5:31. Doi: 10.1186/1477-7525-5-31
- ↑ Emery AE. Мышечные дистрофии. Lancet. 2002 Feb 23;359(9307):687-95